

[ad_1]
Researchers from McMaster College and FAIR Meta have developed a brand new machine studying (ML) approach for orbital-free density purposeful principle (OF-DFT). This ML technique optimizes the entire power perform and efficiently replicates digital density throughout varied chemical techniques. The method has been utilized to simulate lithium hydride, hydrogen, and water molecules, and the memory-efficient gradient optimization technique enhances accuracy by optimizing the Laplacian operator and fixing Hartree and exterior potential functionals.
There are present strategies to calculate molecular digital power, akin to the standard Kohn-Sham density purposeful principle (KS-DFT), which depends on molecular orbitals. Nevertheless, an unexplored method referred to as OF-DFT has been developed that makes use of electron density to reduce a degree and is extra appropriate for complicated techniques.
OF-DFT is an electron density-centric computational method in quantum chemistry and condensed matter physics, providing benefits over KS-DFT for big techniques. It determines ground-state properties via electron density minimization, aligning with the Hohenberg-Kohn theorems. It introduces a novel method utilizing a normalizing circulation ansatz to parameterize and optimize the digital density, efficiently replicating it for numerous chemical techniques.
The proposed technique for optimizing whole power perform in OF-DFT entails using a normalizing circulation ansatz to parameterize digital density throughout varied chemical techniques. It’s achieved via steady normalizing flows that rework digital density by fixing unusual differential equations utilizing a neural community. Gradient-based algorithms are used for whole power optimization, whereas Monte Carlo sampling is utilized for related portions. Additionally, a memory-efficient gradient optimization technique is employed for fixing the Laplacian operator and functionals associated to the Hartree and exterior potentials in OF-DFT.
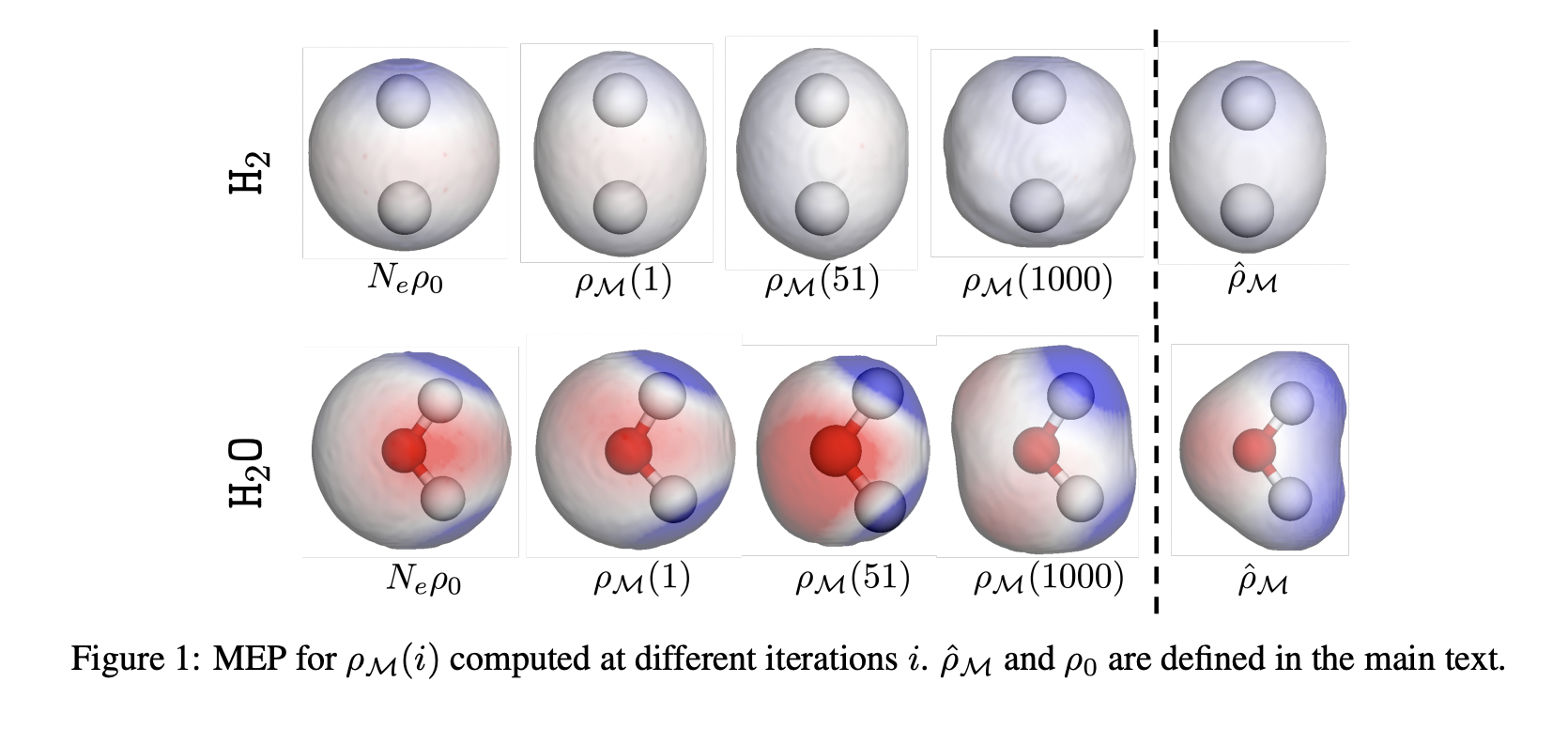
The tactic efficiently modeled diatomic molecules, particularly LiH, and carried out intensive simulations of hydrogen and water molecules. The mannequin precisely replicated digital density in varied chemical techniques, exhibiting adjustments in density and potential power floor in the course of the optimization of H2 and H2O molecules. Comparative evaluation with the Hartree-Fock mannequin utilizing the STO-3G foundation set demonstrated larger density round nuclei within the steady normalizing circulation mannequin. The density purposeful worth was computed utilizing an exponential transferring common all through the optimization course of.
In conclusion, the OF-DFT method using steady normalizing flows for density transformation is a promising constraint-free answer for precisely describing digital density and potential power surfaces throughout varied chemical techniques. Its means to duplicate excessive density round nuclei, as demonstrated within the examine with molecules akin to LiH, hydrogen, and water, highlights its potential for additional refinement and utility.
Future work in OF-DFT digital construction calculations may contain:
- Refining the normalizing circulation ansatz for digital density.
- Extending the continual normalizing circulation method to extra complicated chemical techniques.
- Conducting comparative analyses to evaluate the accuracy of the CNF mannequin.
- Integrating the CNF mannequin with different machine studying strategies to enhance effectivity and precision.
Take a look at the Paper. All credit score for this analysis goes to the researchers of this challenge. Additionally, don’t neglect to hitch our 33k+ ML SubReddit, 41k+ Facebook Community, Discord Channel, and Email Newsletter, the place we share the most recent AI analysis information, cool AI tasks, and extra.
If you like our work, you will love our newsletter..
Whats up, My identify is Adnan Hassan. I’m a consulting intern at Marktechpost and shortly to be a administration trainee at American Categorical. I’m at present pursuing a twin diploma on the Indian Institute of Know-how, Kharagpur. I’m captivated with expertise and need to create new merchandise that make a distinction.
[ad_2]
Source link
