

[ad_1]
The historical past of every dwelling being is written in its genome, which is saved as DNA and current in practically each cell of the physique. No two cells are the identical, even when they share the identical DNA and cell kind, as they nonetheless differ within the regulators that management how DNA is expressed by the cell. The human genome consists of three billion base pairs unfold over 23 chromosomes. Inside this huge genetic code, there are roughly 20,000 to 25,000 genes, constituting the protein-coding DNA and accounting for about 1% of the whole genome [1]. To discover the functioning of advanced programs in our our bodies, particularly this small coding portion of DNA, a exact sequencing technique is critical, and single-cell sequencing (sc-seq) expertise matches this objective.
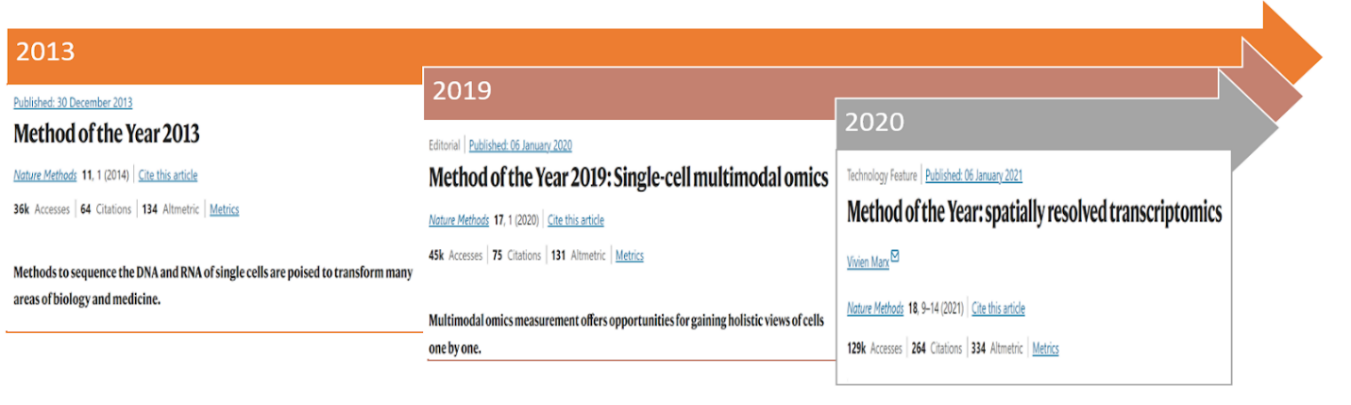
In 2013, Nature chosen single-cell RNA sequencing because the Methodology of the 12 months [2] (Determine 3), highlighting the significance of this technique for exploring mobile heterogeneity by means of the sequencing of DNA and RNA on the particular person cell degree. Subsequently, quite a few instruments have emerged for the evaluation of single-cell RNA sequencing information. For instance, the scRNA-tools database has been compiling software program for the evaluation of single-cell RNA information since 2016, and by 2021, the database consists of over 1000 instruments [3]. Amongst these instruments, many contain strategies that leverage Deep Studying methods, which would be the focus of this text – we’ll discover the pivotal function that Deep Studying, specifically, has performed as a key enabler for advancing single-cell sequencing applied sciences.
Background
Circulate of genetic data from DNA to protein in cells
Let’s first go over what precisely cells and sequences are. The cell is the basic unit of our our bodies and the important thing to understanding how our our bodies operate in good well being and the way molecular dysfunction results in illness. Our our bodies are fabricated from trillions of cells, and practically each cell comprises three genetic data layers: DNA, RNA, and protein. DNA is an extended molecule containing the genetic code that makes every particular person distinctive. Like a supply code, it consists of a number of directions displaying easy methods to make every protein in our our bodies. These proteins are the workhorses of the cell that perform practically each job mandatory for mobile life. For instance, the enzymes that catalyze chemical reactions inside the cell and DNA polymerases that contribute to DNA replication throughout cell division, are all proteins. The cell synthesizes proteins in two steps: Transcription and Translation (Determine 1), that are often known as gene expression. DNA is first transcribed into RNA, then RNA is translated into protein. We are able to contemplate RNA as a messenger between DNA and protein.
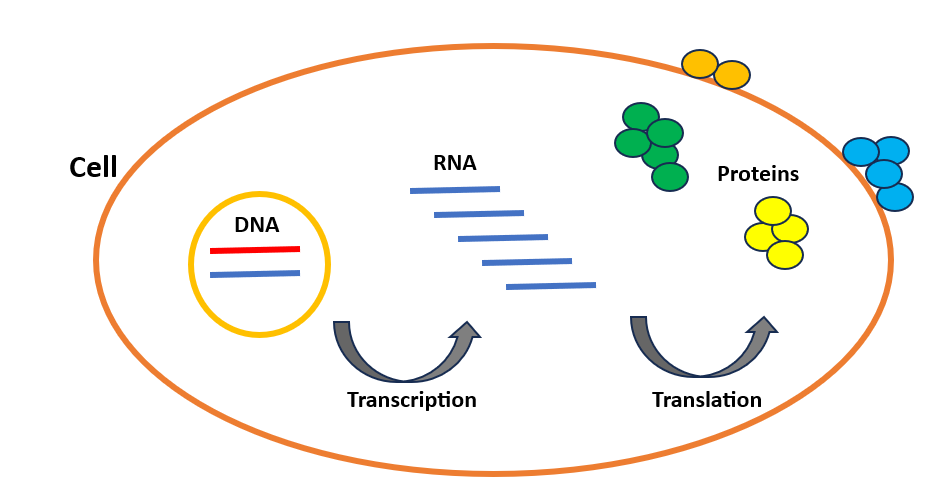
Whereas the cells of our physique share the identical DNA, they fluctuate of their organic exercise. As an illustration, the distinctions between immune cells and coronary heart cells are decided by the genes which might be both activated or deactivated in these cells. Usually, when a gene is activated, it results in the creation of extra RNA copies, leading to elevated protein manufacturing. Due to this fact, as cell sorts differ primarily based on the amount and sort of RNA/protein molecules synthesized, it turns into intriguing to evaluate the abundance of those molecules on the single-cell degree. This can allow us to research the conduct of our DNA inside every cell and attain a high-resolution perspective of the varied elements of our our bodies.
Typically, all single-cell sequencing applied sciences might be divided into three important steps:
- Isolation of single cells from the tissue of curiosity and extraction of genetic materials from every remoted cell
- Amplification of genetic materials from every remoted cell and library preparation
- Sequencing of the library utilizing a next-generation sequencer and information evaluation
Navigating by means of the intricate steps of mobile biology and single-cell sequencing applied sciences, a pivotal query emerges: How is single-cell sequencing information represented numerically?
Construction of single-cell sequencing information
The construction of single-cell sequencing information takes the type of a matrix (Determine 2), the place every row corresponds to a cell that has been sequenced and annotated with a novel barcode. The variety of rows equals the whole variety of cells analyzed within the experiment. Then again, every column corresponds to a selected gene. Genes are the practical models of the genome that encode directions for the synthesis of proteins or different practical molecules. Within the case of scRNA seq information, the numerical entries within the matrix symbolize the expression ranges of genes in particular person cells. These values point out the quantity of RNA produced from every gene in a selected cell, offering insights into the exercise of genes inside completely different cells.

Single Cell Sequencing Overview
For greater than 150 years, biologists have needed to establish all of the cell sorts within the human physique and classify them into distinct sorts primarily based on correct descriptions of their properties. The Human Cell Atlas Undertaking (HCAP), the genetic equal of the Human Genome Undertaking [4], is a world collaborative effort to map all of the cells within the human physique.” We are able to conceptualize the Human Cell Atlas as a map endeavoring to painting the human physique coherently and systematically. Very like Google Maps, which permits us to zoom in for a better examination of intricate particulars, the Human Cell Atlas offers insights into spatial data, inside attributes, and even the relationships amongst parts”, explains Aviv Regev, a computational and programs biologist on the Broad Institute of MIT and Harvard and Government Vice President and Head of Genentech Analysis.
This analogy seamlessly aligns with the broader impression of single-cell sequencing, because it permits the evaluation of particular person cells as a substitute of bulk populations. This expertise proves invaluable in addressing intricate organic inquiries associated to developmental processes and comprehending heterogeneous mobile or genetic modifications below numerous therapy situations or illness states. Moreover, it facilitates the identification of novel cell sorts inside a given mobile inhabitants. The initiation of the primary single-cell RNA sequencing (scRNA-seq) paper in 2009 [5], subsequently designated because the “technique of the yr” in 2013 [2], marked the genesis of an in depth endeavor to advance each experimental and computational methods devoted to unraveling the intricacies of single-cell transcriptomes.
Because the technological panorama evolves, the narrative transitions to the developments in single-cell analysis, notably the early concentrate on single-cell RNA sequencing (scRNA-seq) resulting from its cost-effectiveness in learning advanced cell populations.” In some methods, RNA has at all times been one of many best issues to measure,” says Satija [6], a researcher on the New York Genome Middle (NYGC). But, the speedy improvement of single-cell expertise has ushered in a brand new period of potentialities—multimodal single-cell information integration. Acknowledged because the “Methodology of the 12 months 2019” by Nature [7] (Determine 3), this method permits the measurement of various mobile modalities, together with the genome, epigenome, and proteome, inside the identical cell. The layering of a number of items of knowledge offers highly effective insights into mobile identification, posing the problem of successfully modeling and mixing datasets generated from multimodal measurements. This integration problem is met with the introduction of Multi-view studying [8] strategies, exploring frequent variations throughout modalities. This subtle method, incorporating deep studying methods, showcases related outcomes throughout numerous fields, notably in biology and biomedicine.
Amidst these developments, a definite problem surfaces within the persistent limitation of single-cell RNA sequencing—the lack of spatial data throughout transcriptome profiling by isolating cells from their authentic place. Spatially resolved transcriptomics (SRT) emerges as a pivotal answer [9], addressing the problem by preserving spatial particulars throughout the examine of advanced organic programs. This recognition of spatially resolved transcriptomics as the tactic of the yr 2020 solidifies its place as a essential answer to the challenges inherent in advancing our understanding of advanced organic programs.
Having explored the panorama of single-cell sequencing, allow us to now delve into the function of deep studying within the context of single-cell sequencing.
Deep Studying on single-cell sequencing
Deep studying is more and more employed in single-cell evaluation resulting from its capability to deal with the complexity of single-cell sequencing information. In distinction, typical machine-learning approaches require vital effort to develop a function engineering technique, usually designed by area consultants. The deep studying method, nevertheless, autonomously captures related traits from single-cell sequencing information, addressing the heterogeneity between single-cell sequencing experiments, in addition to the related noise and sparsity in such information. Under are three key causes for the applying of deep studying in single-cell sequencing:
- Excessive-Dimensional Information: Single-cell sequencing generates high-dimensional information, with hundreds of genes and their expression ranges measured for every cell. Deep studying fashions are adept at capturing advanced relationships and patterns inside this information, which might be difficult for conventional statistical strategies.
- Non-Linearity: Single-cell gene expression information is characterised by its inherent nonlinearity between gene expressions and cell-to-cell heterogeneity. Conventional statistical strategies encounter difficulties in capturing the non-linear relationships current in single-cell gene expression information. In distinction, deep studying fashions are versatile and capable of be taught advanced non-linear mappings.
- Heterogeneity: Single-cell information is usually characterised by various cell populations with various gene expression profiles, presenting a fancy panorama. Deep studying fashions can play an important function in figuring out, clustering, and characterizing these distinct cell sorts or subpopulations, thereby facilitating a deeper understanding of mobile heterogeneity inside a pattern.
As we discover the explanations behind utilizing deep studying in single-cell sequencing information, it leads us to the query: What deep studying architectures are sometimes utilized in sc-seq information evaluation?
Background on Autoencoders
Autoencoders (AEs) stand out amongst numerous deep-learning architectures (comparable to GANs and RNNs) as an particularly relied upon technique for decoding the complexities of single-cell sequencing information. Broadly employed for dimensionality discount whereas preserving the inherent heterogeneity within the single-cell sequencing information. By clustering cells within the reduced-dimensional area generated by autoencoders, researchers can successfully establish and characterize completely different cell sorts or subpopulations. This method enhances our skill to discern and analyze the various mobile elements inside single-cell datasets. In distinction to non-deep studying fashions, comparable to principal part evaluation (PCA), that are integral elements of established scRNA-seq information evaluation software program like Seurat [10], autoencoders distinguish themselves by uncovering non-linear manifolds. Whereas PCA is constrained to linear transformations, the flexibleness of autoencoders to seize advanced non-linear mappings makes it a sophisticated technique to search out nuanced relationships embedded in single-cell genomics.
To mitigate the overfitting problem related to autoencoders, a number of enhancements to the autoencoder construction have been applied, particularly tailor-made to supply benefits within the context of sc-seq information. One notable adaptation typically used within the context of sc-seq information is the denoising autoencoder (DAEs), which amplifies the autoencoder’s reconstruction functionality by introducing noise to the preliminary community layer. This includes randomly remodeling a few of its models to zero. The Denoising Autoencoder then reconstructs the enter from this deliberately corrupted model, empowering the community to seize extra related options and stopping it from merely memorizing the enter (overfitting). This refinement considerably bolsters the mannequin’s resilience in opposition to information noise, thereby elevating the standard of the low-dimensional illustration of samples (i.e., bottleneck) derived from the sc-seq information.
A 3rd variation of autoencoders continuously employed in sc-seq information evaluation is variational autoencoders (VAEs), exemplified by fashions like scGen [19], scVI [14], scANVI [28], and so forth. VAEs, as a kind of generative mannequin, be taught a latent illustration distribution of the info. As a substitute of encoding the info right into a vector of p-dimensional latent variables, the info is encoded into two vectors of dimension p: a vector of means η and a vector of ordinary deviations σ. VAEs introduce a probabilistic ingredient to the encoding course of, facilitating the era of artificial single-cell information and providing insights into the range inside a cell inhabitants. This nuanced method provides one other layer of complexity and richness to the exploration of single-cell genomics.
Functions of deep studying in sc-seq information evaluation
This part outlines the principle functions of deep studying in bettering numerous levels of sc-seq information evaluation, highlighting its effectiveness in advancing essential facets of the method.
scRNA-seq information imputation and denoising
Single-cell RNA sequencing (scRNA-seq) information encounter inherent challenges, with dropout occasions being a distinguished concern that results in vital points—leading to sparsity inside the gene expression matrix, typically characterised by a considerable variety of zero values. This sparsity considerably shapes downstream bioinformatics analyses. Many of those zero values come up artificially resulting from deficiencies in sequencing methods, together with issues like insufficient gene expression, low seize charges, sequencing depth, or different technical elements. As a consequence, the noticed zero values don’t precisely mirror the true underlying expression ranges. Therefore, not all zeros in scRNA-seq information might be thought of mere lacking values, deviating from the traditional statistical method of imputing lacking information values. Given the intricate distinction between true and false zero counts, conventional imputation strategies with predefined lacking values could show insufficient for scRNA-seq information. As an illustration, a classical imputation technique, like Imply Imputation, would possibly entail substituting these zero values with the common expression degree of that gene throughout all cells. Nonetheless, this method runs the danger of oversimplifying the complexities launched by dropout occasions in scRNA-seq information, doubtlessly resulting in biased interpretations.
ScRNA-seq information imputation strategies might be divided into two classes: deep studying–primarily based imputation technique and non–deep studying imputation technique. The non–deep studying imputation algorithms contain becoming statistical likelihood fashions or using the expression matrix for smoothing and diffusion. This simplicity renders it efficient for sure forms of samples. For instance, Wagner et al. [11] utilized the k-nearest neighbors (KNN) technique, figuring out nearest neighbors between cells and aggregating gene-specific Distinctive Molecular Identifiers (UMI) counts to impute the gene expression matrix. In distinction, Huang et al. [12] proposed the SVAER algorithm, leveraging gene-to-gene relationships for imputing the gene expression matrix. For bigger datasets (comprising tens of hundreds or extra), high-dimensional, sparse, and complicated scRNA-seq information, conventional computational strategies face difficulties, typically rendering evaluation utilizing these strategies tough and infeasible. Consequently, many researchers have turned to designing strategies primarily based on deep studying to handle these challenges.
Most deep studying algorithms for imputing dropout occasions are primarily based on autoencoders (AEs). As an illustration, in 2018, Eraslan et al. [13] launched the deep depend autoencoder (DCA). DCA makes use of a deep autoencoder structure to handle dropout occasions in single-cell RNA sequencing (scRNA-seq) information. It incorporates a probabilistic layer within the decoder to mannequin the dropout course of. This probabilistic layer accommodates the uncertainty related to dropout occasions, enabling the mannequin to generate a distribution of attainable imputed values. To seize the traits of depend information in scRNA-seq, DCA fashions the noticed counts as originating from a destructive binomial distribution.
Single-cell variational inference (scVI) is one other deep studying algorithm launched by Lopez et al. [14]. ScVI is a probabilistic variational autoencoder (VAE) that mixes deep studying and probabilistic modeling to seize the underlying construction of the scRNA-seq information. ScVI can be utilized for imputation, denoising, and numerous different duties associated to the evaluation of scRNA-seq information. In distinction to the DCA mannequin, scVI employs Zero-Inflated Adverse Binomial (ZINB) distribution within the decoder half to generate a distribution of attainable counts for every gene in every cell. The Zero-Inflated Adverse Binomial (ZINB) distribution permits modeling the likelihood of a gene expression being zero (to mannequin dropout occasions) in addition to the distribution of optimistic values (to mannequin non-zero counts).
Moreover, one other examine addressed the scRNA-seq information imputation problem by introducing a recurrent community layer of their mannequin, often known as scScope [15]. This novel structure iteratively performs imputations on zero-valued entries of enter scRNA-seq information. The pliability of scScope’s design permits for the iterative enchancment of imputed outputs by means of a selected variety of recurrent steps (T). Noteworthy is the truth that decreasing the time recurrence of scScope to 1 (i.e., T = 1) transforms the mannequin into a conventional autoencoder (AE). As scScope is actually a modification of conventional AEs, its runtime is corresponding to different AE-based fashions.
It is vital to notice that the applying of deep studying in scRNA-seq information imputation and denoising is especially advantageous resulting from its skill to seize non-linear relationships amongst genes. This contrasts with normal linear approaches, making deep studying more proficient at offering knowledgeable and correct imputation methods within the context of single-cell genomics.
Batch impact elimination
Single-cell information is usually aggregated from various experiments that change when it comes to experimental laboratories, protocols, pattern compositions, and even expertise platforms. These variations end in vital variations or batch results inside the information, posing a problem within the evaluation of organic variations of curiosity throughout the course of of knowledge integration. To deal with this challenge, it turns into essential to appropriate batch results by eradicating technical variance when integrating cells from completely different batches or research. The primary technique that seems for batch correction is a linear technique primarily based on linear regression comparable to Limma package deal [16] that gives the removeBatchEffect operate which inserts a linear mannequin that considers the batches and their impression on gene expression. After becoming the mannequin, it units the coefficients related to every batch to zero, successfully eradicating their impression. One other technique referred to as ComBat [17] does one thing related however provides an additional step to refine the method, making the correction much more correct by utilizing a method referred to as empirical Bayes shrinkage.
Nonetheless, batch results might be extremely nonlinear, making it tough to appropriately align completely different datasets whereas preserving key organic variations. In 2018, Haghverdi et al. launched the Mutual Nearest Neighbors (MNN) algorithm to establish pairs of cells from completely different batches in single-cell information [18]. These recognized mutual nearest neighbors help in estimating batch results between batches. By making use of this correction, the gene expression values are adjusted to account for the estimated batch results, aligning them extra carefully and decreasing discrepancies launched by the completely different batches. For intensive single-cell datasets with extremely nonlinear batch results, conventional strategies could show much less efficient, prompting researchers to discover the applying of neural networks for improved batch correction.
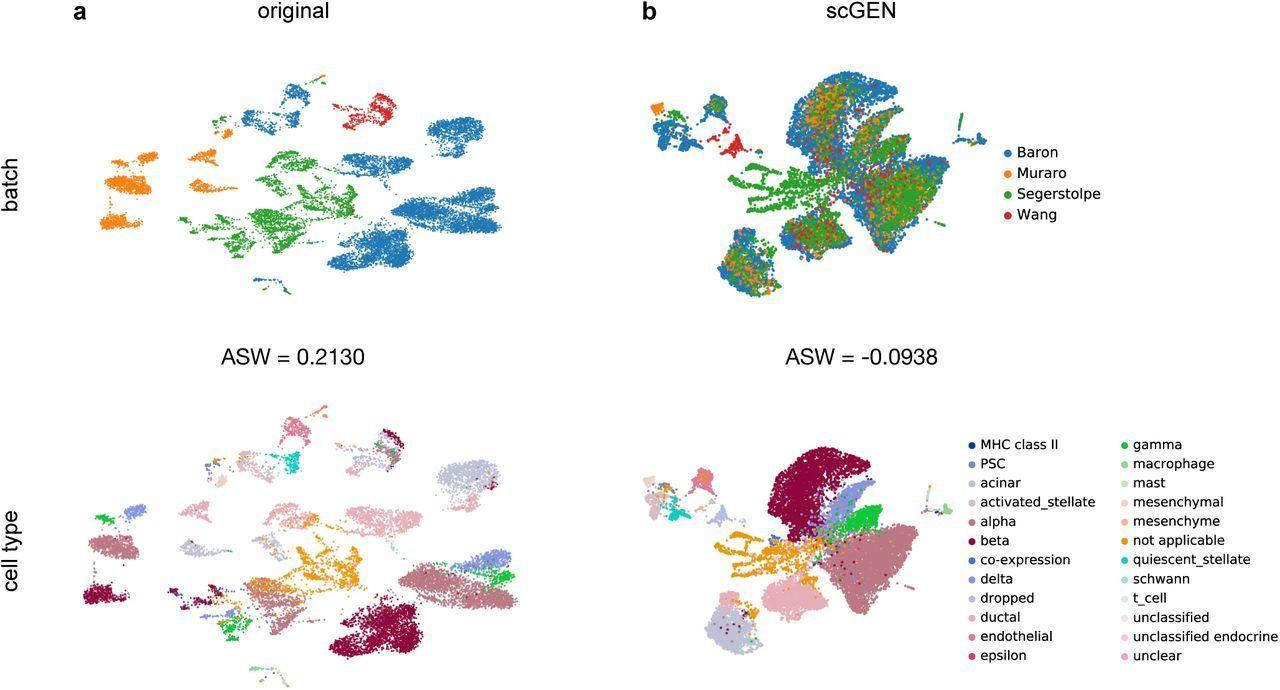
One of many pioneering fashions that make use of deep studying for batch correction is the scGen mannequin. Developed by Lotfollahi et al., ScGen [19] makes use of a variational autoencoder (VAE) structure. This includes pre-training a VAE mannequin on a reference dataset to regulate actual single-cell information and alleviate batch results. Initially, the VAE is skilled to seize latent options inside the reference dataset’s cells. Subsequently, this skilled VAE is utilized to the precise information, producing latent representations for every cell. The adjustment of gene expression profiles is then primarily based on aligning these latent representations, to scale back batch results and harmonize profiles throughout completely different experimental situations.
Then again, Zou et al. launched DeepMNN [20], which employs a residual neural community and the mutual nearest neighbor (MNN) algorithm for scRNA-seq information batch correction. Initially, MNN pairs are recognized throughout batches in a principal part evaluation (PCA) subspace. Subsequently, a batch correction community is constructed utilizing two stacked residual blocks to take away batch results. The loss operate of DeepMNN includes a batch loss, computed primarily based on the gap between cells in MNN pairs within the PCA subspace, and a weighted regularization loss, guaranteeing the community’s output similarity to the enter.
Nearly all of current scRNA-seq strategies are designed to take away batch results first after which cluster cells, which doubtlessly overlooks sure uncommon cell sorts. Not too long ago, Xiaokang et al. developed scDML [21], a deep metric studying mannequin to take away batch impact in scRNA-seq information, guided by the preliminary clusters and the closest neighbor data intra and inter-batches. First, the graph-based clustering algorithm is used to group cells primarily based on gene expression similarities, then the KNN algorithm is utilized to establish k-nearest neighbors for every cell within the dataset, and the MNN algorithm to establish mutual nearest neighbors, specializing in reciprocal relationships between cells. To take away batch results, deep triplet studying is employed, contemplating arduous triplets. This helps in studying a low-dimensional embedding that accounts for the unique high-dimensional gene expression and removes batch results concurrently.
Cell kind annotation
Cell kind annotation in single-cell sequencing includes the method of figuring out and labeling particular person cells primarily based on their gene expression profiles, which permits researchers to seize the range inside a heterogeneous inhabitants of cells, and perceive the mobile composition of tissues, and the practical roles of various cell sorts in organic processes or ailments. Historically, researchers have used guide strategies [22] to annotate cell sub-populations. This includes figuring out gene markers or gene signatures which might be differentially expressed in a selected cell cluster. As soon as gene markers are recognized, researchers manually interpret the organic relevance of those markers to assign cell-type labels to the clusters. This conventional guide annotation method is time-consuming and requires appreciable human effort, particularly when coping with large-scale single-cell datasets. As a result of challenges related to guide annotation, researchers are turning to automate and streamline the cell annotation course of.
Two major methods are employed for cell kind annotation: unsupervised-based and supervised-based. Within the unsupervised realm, clustering strategies comparable to Scanpy [23] and Seurat [10] are utilized, demanding prior data of established mobile markers. The identification of clusters hinges on the unsupervised grouping of cells with out exterior reference data. Nonetheless, a downside to this method is a possible lower in replicability with an elevated variety of clusters and a number of choices of cluster marker genes.
Conversely, supervised-based methods depend on deep-learning fashions skilled on labeled information. These fashions discern intricate patterns and relationships inside gene expression information throughout coaching, enabling them to foretell cell sorts for unlabeled information primarily based on acquired patterns. For instance, Joint Integration and Discrimination (JIND) [24] deploys a GAN-style deep structure, the place an encoder is pre-trained on classification duties, circumventing the necessity for an autoencoder framework. This mannequin additionally accounts for batch results. AutoClass [25] integrates an autoencoder and a classifier, combining output reconstruction loss with a classification loss for cell annotation alongside information imputation. Moreover, TransCluster, [26] rooted within the Transformer framework and convolutional neural community (CNN), employs function extraction from the gene expression matrix for single-cell annotation.
Regardless of the facility of deep neural networks, acquiring a lot of precisely and unbiasedly annotated cells for coaching is difficult, given the labor-intensive guide inspection of marker genes in scRNAseq information. In response, semi-supervised studying has been leveraged in computational cell annotation. As an illustration, the SemiRNet [27] mannequin makes use of each unlabeled and a restricted quantity of labeled scRNAseq cells to implement cell identification. SemiRNet, primarily based on recurrent convolutional neural networks (RCNN), incorporates a shared community, a supervised community, and an unsupervised community. Moreover, single‐cell ANnotation utilizing Variational Inference (scANVI) [28], a semi‐supervised variant of scVI [14], maximizes the utility of current cell state annotations. Cell BLAST, an autoencoder-based generative mannequin, harnesses large-scale reference databases to be taught nonlinear low-dimensional representations of cells, using a complicated cell similarity metric—normalized projection distance—to map question cells to particular cell sorts and establish novel cell sorts.
Multi-omics Information Integration
Current research have demonstrated the potential of deep studying fashions in addressing advanced and multimodal organic challenges [29]. Among the many algorithms proposed to date, it’s primarily deep learning-based fashions that present the important computational adaptability mandatory for successfully modeling and incorporating practically any type of omic information together with genomics (learning DNA sequences and genetic variations), epigenomics (inspecting modifications in gene exercise unrelated to DNA sequence, comparable to DNA modifications and chromatin construction), transcriptomics (investigating RNA molecules and gene expression by means of RNA sequencing), and proteomics (analyzing all proteins produced by an organism, together with buildings, abundances, and modifications). Deep Studying architectures, together with autoencoders (AE) and generative adversarial networks (GAN), have been typically utilized in multi-omics integration issues in single cells. The important thing query in multi-omics integration revolves round easy methods to successfully symbolize the various multi-omics information inside a unified latent area.
One of many early strategies developed utilizing Variational Autoencoders (VAE) for the combination of multi-omics single-cell information is named totalVI [30]. The totalVI mannequin, which is VAE-based, provides an answer for successfully merging scRNA-seq and protein information. On this mannequin, totalVI takes enter matrices containing scRNA-seq and protein depend information. Particularly, it treats gene expression information as sampled from a destructive binomial distribution, whereas protein information are handled as sampled from a mix mannequin consisting of two destructive binomial distributions. The mannequin first learns shared latent area representations by means of its encoder, that are then utilized to reconstruct the unique information, making an allowance for the variations between the 2 authentic information modalities. Lastly, the decoder part estimates the parameters of the underlying distributions for each information modalities utilizing the shared latent illustration.
Then again, Zuo et al. [31] launched scMVAE as a multimodal variational autoencoder designed to combine transcriptomic and chromatin accessibility information in the identical particular person cells. scMVAE employs two separate single-modal encoders and two single-modal decoders to successfully mannequin each transcriptomic and chromatin information. It achieves this by combining three distinct joint-learning methods with a probabilistic Gaussian Combination Mannequin.
Not too long ago, Lotfollahi et al. [32] launched an unsupervised deep generative mannequin often known as MULTIGRATE for the combination of multi-omic datasets. MULTIGRATE employs a multi-modal variational autoencoder construction that shares some similarities with the scMVAE mannequin. Nonetheless, it provides added generality and the aptitude to combine each paired and unpaired single-cell information. To reinforce cell alignment, the loss operate incorporates Most Imply Discrepancy (MMD), penalizing any misalignment between the purpose clouds related to completely different assays. Incorporating switch studying, MULTIGRATE can map new multi-omic question datasets right into a reference atlas and in addition carry out imputations for lacking modalities.
Conclusion
The appliance of deep studying in single-cell sequencing capabilities as a sophisticated microscope, revealing intricate insights inside particular person cells and offering a profound understanding of mobile heterogeneity and complexity in organic programs. This cutting-edge expertise empowers scientists to discover beforehand undiscovered facets of mobile conduct. Nonetheless, the problem lies in selecting between conventional instruments and the plethora of obtainable deep-learning choices. The panorama of instruments is huge, and researchers should fastidiously contemplate elements comparable to information kind, complexity, and the precise organic questions at hand. Navigating this decision-making course of requires a considerate analysis of the strengths and limitations of every device in relation to analysis objectives.
Then again, a essential want within the improvement of deep studying approaches for single-cell RNA sequencing (scRNA-seq) evaluation is strong benchmarking. Whereas many research evaluate deep studying efficiency to straightforward strategies, there’s a lack of complete comparisons throughout numerous deep studying fashions. Furthermore, strategies typically declare superiority primarily based on particular datasets and tissues (e.g., pancreas cells, immune cells), making it difficult to judge the need of particular phrases or preprocessing steps. Addressing these challenges requires an understanding of when deep studying fashions fail and their limitations. Recognizing which forms of deep studying approaches and mannequin buildings are helpful in particular circumstances is essential for growing new approaches and guiding the sphere.
Within the realm of multi-omics single-cell integration, most deep studying strategies intention to discover a shared latent illustration for all modalities. Nonetheless, shared illustration studying faces challenges comparable to heightened noise, sparsity, and the intricate job of balancing modalities. Inherent biases throughout establishments complicate generalization. Regardless of being much less prevalent than single-modality approaches, integrating various modalities with distinctive cell populations is essential. Aims embrace predicting expression throughout modalities and figuring out cells in related states. Regardless of developments, additional efforts are important for enhanced efficiency, notably regarding distinctive or uncommon cell populations current in a single expertise however not the opposite.
Creator Bio
Fatima Zahra El Hajji holds a grasp’s diploma in bioinformatics from the Nationwide Faculty of Laptop Science and Methods Evaluation (ENSIAS), she subsequently labored as an AI intern at Piercing Star Applied sciences. Presently, she is a Ph.D. scholar on the College Mohammed VI Polytechnic (UM6P), working below the supervision of Dr. Rachid El Fatimy and Dr. Tariq Daouda. Her analysis focuses on the applying of deep studying methods in single-cell sequencing information.
Quotation
For attribution in educational contexts or books, please cite this work as
Fatima Zahra El Hajji, "Deep studying for single-cell sequencing: a microscope to see the range of cells", The Gradient, 2024.
BibTeX quotation:
@article{elhajji2023nar,
creator = {El Hajji, Fatima Zahra},
title = {Deep studying for single-cell sequencing: a microscope to see the range of cells},
journal = {The Gradient},
yr = {2024},
howpublished = {url{deep-learning-for-single-cell-sequencing-a-microscope-to-uncover-the-rich-diversity-of-individual-cells},
}References
- Nationwide Human Genome Analysis Institute (NHGRI) : A Temporary Information to Genomics , https://www.genome.gov/about-genomics/fact-sheets/A-Brief-Guide-to-Genomics
- Methodology of the 12 months 2013. Nat Strategies 11, 1 (2014). https://doi.org/10.1038/nmeth.2801
- Zappia, L., Theis, F.J. Over 1000 instruments reveal traits within the single-cell RNA-seq evaluation panorama. Genome Biol 22, 301 (2021). https://doi.org/10.1186/s13059-021-02519-4
- Collins FS, Fink L. The Human Genome Undertaking. Alcohol Well being Res World. 1995;19(3):190-195. PMID: 31798046; PMCID: PMC6875757.
- Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome evaluation of a single cell. Nat Strategies. 2009; 6: 377-382.
- Eisenstein, M. The key lifetime of cells. Nat Strategies 17, 7–10 (2020). https://doi.org/10.1038/s41592-019-0698-y
- Methodology of the 12 months 2019: Single-cell multimodal omics. Nat Strategies 17, 1 (2020). https://doi.org/10.1038/s41592-019-0703-5
- Zhao, Jing et al. “Multi-view studying overview: Current progress and new challenges.” Inf. Fusion 38 (2017): 43-54.
- Zhu, J., Shang, L. & Zhou, X. SRTsim: spatial sample preserving simulations for spatially resolved transcriptomics. Genome Biol 24, 39 (2023).
- Butler, A., Hoffman, P., Smibert, P., Papalexi, E., & Satija, R. (2018). Integrating single-cell transcriptomic information throughout completely different situations, applied sciences, and species. Nature biotechnology, 36(5), 411-420
- Wagner, F., Yan, Y., & Yanai, I. (2018). Ok-nearest neighbor smoothing for high-throughput single-cell RNA-Seq information. bioRxiv, 217737. Chilly Spring Harbor Laboratory. https://doi.org/10.1101/217737
- Huang, M., Wang, J., Torre, E. et al. SAVER: gene expression restoration for single-cell RNA sequencing. Nat Strategies 15, 539–542 (2018). https://doi.org/10.1038/s41592-018-0033-z
- Eraslan G, Simon LM, Mircea M, Mueller NS, Theis FJ. Single-cell RNA-seq denoising utilizing a deep depend autoencoder. Nat Commun. 2019 Jan 23;10(1):390. doi: 10.1038/s41467-018-07931-2. PMID: 30674886; PMCID: PMC6344535.
- Lopez, R., Regier, J., Cole, M. B., Jordan, M. I.,& Yosef, N. (2018). Deep generative modeling for single-cell transcriptomics. Nature strategies, 15(12), 1053-1058.
- Y. Deng, F. Bao, Q. Dai, L.F. Wu, S.J. Altschuler Scalable evaluation of cell-type composition from single-cell transcriptomics utilizing deep recurrent studying
- Ritchie ME, Phipson B, Wu D, Hu Y, Regulation CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray research. Nucleic Acids Res. 2015 Apr 20;43(7):e47. doi: 10.1093/nar/gkv007. Epub 2015 Jan 20. PMID: 25605792; PMCID: PMC4402510.
- Johnson W.E. , Li C., Rabinovic A. Adjusting batch results in microarray expression information utilizing empirical bayes strategies. Biostatistics. 2007; 8:118–127.
- Haghverdi, L., Lun, A., Morgan, M. et al. Batch results in single-cell RNA-sequencing information are corrected by matching mutual nearest neighbors. Nat Biotechnol 36, 421–427 (2018). https://doi.org/10.1038/nbt.4091
- Lotfollahi, M., Wolf, F. A., & Theis, F. J. (2019). scGen predicts single-cell perturbation responses. Nature strategies, 16(8), 715-721.
- Zou, B., Zhang, T., Zhou, R., Jiang, X., Yang, H., Jin, X., & Bai, Y. (2021). deepMNN: deep learning-based single-cell RNA sequencing information batch correction utilizing mutual nearest neighbors. Frontiers in Genetics, 1441.
- Yu, X., Xu, X., Zhang, J. et al. Batch alignment of single-cell transcriptomics information utilizing deep metric studying. Nat Commun 14, 960 (2023). https://doi.org/10.1038/s41467-023-36635-5
- Z.A. Clarke, T.S. Andrews, J. Atif, D. Pouyabahar, B.T. Innes, S.A. MacParland, et al. Tutorial: tips for annotating single-cell transcriptomic maps utilizing automated and guide strategies Nat Protoc, 16 (2021), pp. 2749-2764
- Wolf, F., Angerer, P. & Theis, F. SCANPY: large-scale single-cell gene expression information evaluation. Genome Biol 19, 15 (2018). https://doi.org/10.1186/s13059-017-1382-0
- Mohit Goyal, Guillermo Serrano, Josepmaria Argemi, Ilan Shomorony, Mikel Hernaez, Idoia Ochoa, JIND: joint integration and discrimination for automated single-cell annotation, Bioinformatics, Quantity 38, Concern 9, March 2022, Pages 2488–2495, https://doi.org/10.1093/bioinformatics/btac140
- H. Li, C.R. Brouwer, W. Luo A common deep neural community for in-depth cleansing of single-cell RNA-seq information Nat Commun, 13 (2022), p. 1901
- Track T, Dai H, Wang S, Wang G, Zhang X, Zhang Y and Jiao L (2022) TransCluster: A Cell-Sort Identification Methodology for single-cell RNA-Seq information utilizing deep studying primarily based on transformer. Entrance. Genet. 13:1038919. doi: 10.3389/fgene.2022.1038919
- Dong X, Chowdhury S, Victor U, Li X, Qian L. Semi-Supervised Deep Studying for Cell Sort Identification From Single-Cell Transcriptomic Information. IEEE/ACM Trans Comput Biol Bioinform. 2023 Mar-Apr;20(2):1492-1505. doi: 10.1109/TCBB.2022.3173587. Epub 2023 Apr 3. PMID: 35536811.
- Xu, C., Lopez, R., Mehlman, E., Regier, J., Jordan, M. I., & Yosef, N. (2021). Probabilistic harmonization and annotation of single‐cell transcriptomics information with deep generative fashions. Molecular Methods Biology, 17(1), e9620. https://doi.org/10.15252/msb.20209620
- Tasbiraha Athaya, Rony Chowdhury Ripan, Xiaoman Li, Haiyan Hu, Multimodal deep studying approaches for single-cell multi-omics information integration, Briefings in Bioinformatics, Quantity 24, Concern 5, September 2023, bbad313, https://doi.org/10.1093/bib/bbad313
- Gayoso, A., Lopez, R., Steier, Z., Regier, J., Streets, A., & Yosef, N. (2019). A Joint Mannequin of RNA Expression and Floor Protein Abundance in Single Cells. bioRxiv, 791947. https://www.biorxiv.org/content/early/2019/10/07/791947.abstract
- Chunman Zuo, Luonan Chen. Deep-joint-learning evaluation mannequin of single cell transcriptome and open chromatin accessibility information. Briefings in Bioinformatics. 2020.
- Lotfollahi, M., Litinetskaya, A., & Theis, F. J. (2022). Multigrate: single-cell multi-omic information integration.bioRxiv.https://www.biorxiv.org/content/early/2022/03/17/2022.03.16.484643
[ad_2]
Source link
