


[ad_1]
The Problem of Drug Discovery
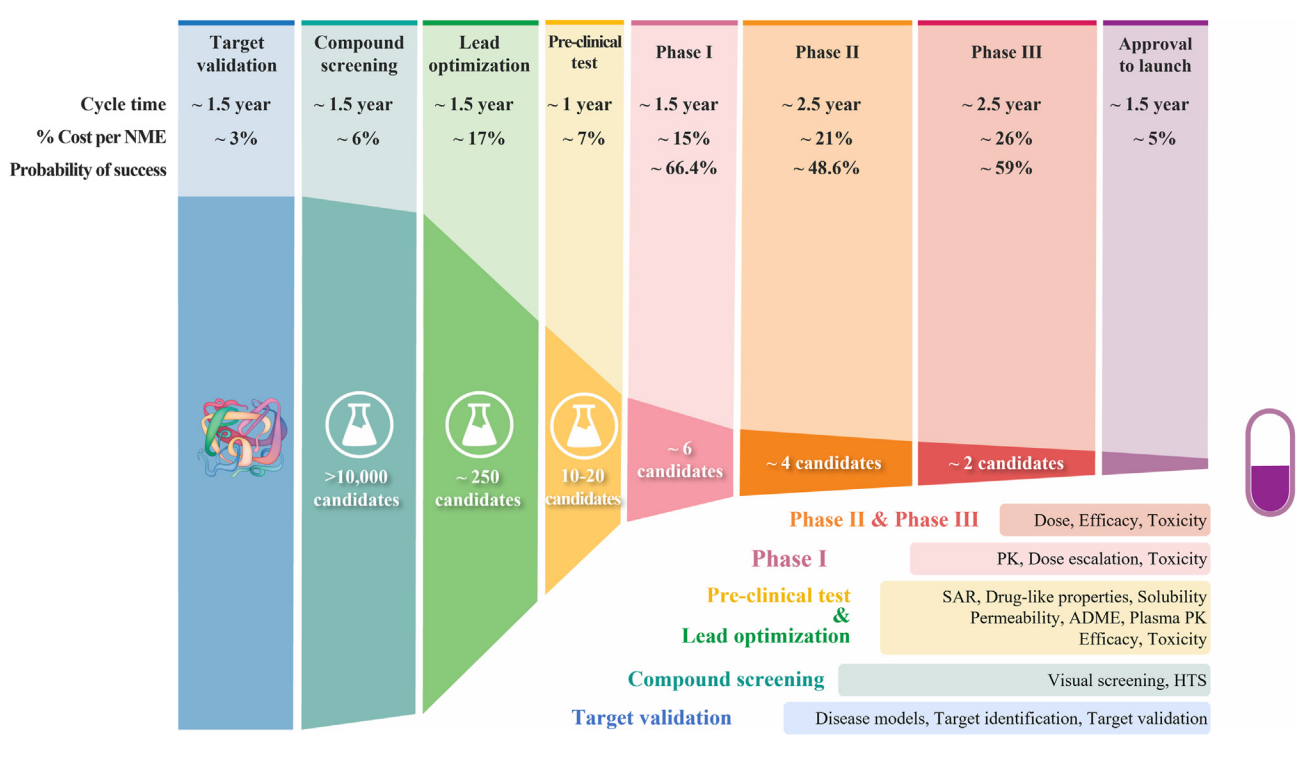
The event of recent medicines is commonly laborious, time-consuming, expensive, and susceptible to failure. The failure fee is notoriously excessive, with the latest statistic being 90% [1,2]. The vary of potential drug-like molecules is estimated to be between $10^{23}$ and $10^{60}$, leading to a big and discrete search house, whereas it’s predicted that “solely” about $10^8$ molecular substances have been synthesised [3]. The chemical house of molecular candidates is huge, and inside this house, attention-grabbing and highly effective medicine are ready to be discovered.
Impactful medicine, like the invention of thalidomide or penicillin, have modified the course of drugs and formed the sector. Nonetheless, the primary drug is infamous for being a part of the thalidomide tragedy, which precipitated beginning defects and deaths amongst pregnant girls within the Sixties [4]. The basis trigger was ignored handedness (or, in jargon, the chirality) between the molecular compounds, i.e. molecules which might be non-superimposable on their mirror photographs with reference to a symmetry axis [5]. A left-handed compound may need totally different pharmacological properties than a right-handed compound; might it yield a toxin, or might it yield the following “surprise drug”?
The examine of symmetry, together with a greater understanding of the functioning of the molecules, is subsequently important to the drug improvement pipeline. Nonetheless, present analysis is predominantly centered on the examine of 1-D and 2-D molecular representations, as a substitute of leveraging the total geometrical info inside these fashions. Growing instruments to leverage the geometrical info and effectively navigate the search house is without doubt one of the core challenges in fashionable chemistry. Thankfully, the sector of geometric deep studying, a subject that exploits the symmetries of machine studying fashions in non-Euclidean domains [6], is more and more getting used to hurry up almost each stage of the drug improvement pipeline. From the prediction of the physicochemical properties of a specific compound to scientific improvement, whereas respecting the geometry of the duty at hand [7] — all of them contribute to addressing the grand challenges going through drug discovery. This text will cowl how the appliance of geometric deep studying and the sector of molecular machine studying is ushering us into a brand new period of scientific discovery.
A Temporary Historical past of Laptop-Aided Drug Discovery
For the previous three many years, computer-aided discovery and design strategies have been a big contributor to the event of comparatively small, therapeutic molecules [8]. Ever since, there was a substantial enhance within the quantity of public quantitative molecular bioactivity knowledge and biomedical knowledge on the consequences of a molecule on a dwelling system [9,10]. Resulting from this steep enhance within the quantity of information, it shortly turns into infeasible to mine information by hand to generate new insights; automated instruments, similar to machine studying algorithms, type one answer to this drawback. Simplified correlations might be inferred between the enter components that retain the required info required to get to the specified end result. This offers room to accommodate extra knowledge, nonetheless, explainability and scientific accuracy (not statistical accuracy) might be compromised.
The usage of deep studying has confirmed to be extremely profitable in data-rich fields similar to laptop imaginative and prescient, pure language processing, and speech recognition [11]. Constructing upon these successes, deep studying is more and more being utilized to issues within the pure sciences, which incorporates the sector of chemistry. Conventional advances in chemistry have mixed the implementation of novel experiments with analytic principle, which mirrors the classical scientific technique by growing falsifiable hypotheses. Now, as we’re steering extra into the period of huge knowledge, computer-aided approaches are step by step being utilized to speed up our scientific analysis and understanding. Equally, these strategies enable for data-driven analysis that generates falsifiable hypotheses that are then examined [12].
Information-driven purposes have more and more been proven to speed up fixing various issues within the drug discovery pipeline [13] — from using predictive analytical strategies in high-throughput screening to using generative fashions for locating and designing novel compounds. Whereas the exploration of the chemical house is useful, it sadly lacks the power to find novel and unknown chemical compounds. The principle cause for that is that the present chemical house, which is merely a fraction of the lively chemical house, is restricted to established, well-known compounds.
One of the vital important advances within the subject of molecular machine studying is the power to foretell the physicochemical properties of particular medicine. The primary method included using Quantitative Construction-Exercise Relationship (QSAR) fashions. These fashions use molecular descriptors to foretell the physicochemical properties and organic actions of molecular compounds in regression and classification duties [14]. By way of advances in synthetic intelligence, the paradigm shifted extra to using deep studying fashions. Classical machine studying strategies, similar to regression fashions, nonetheless outperform deep studying strategies at sure advanced benchmark duties as a result of restricted knowledge availability [15].
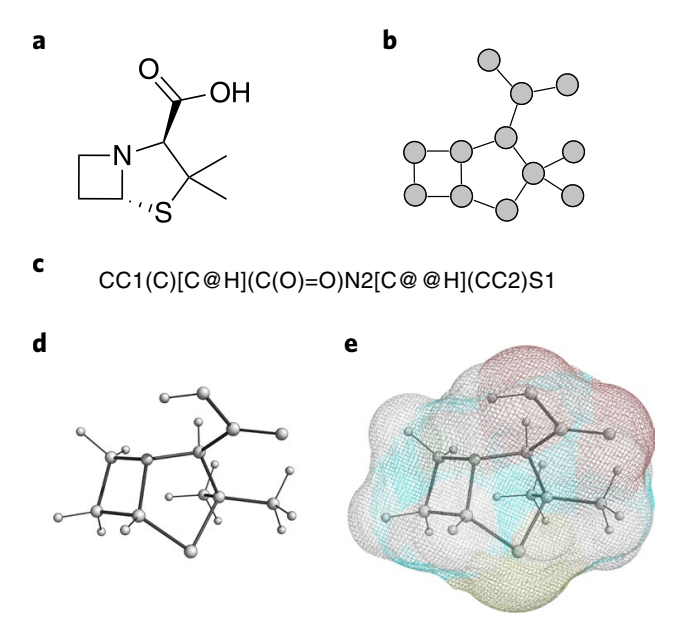
We will signify molecular constructions in a number of distinct methods, as seen in Determine 2. Growing a one-size-fits-all and generalizable answer is a big problem, as these representations seize and mirror totally different legitimate configurations of the identical molecular occasion. To this finish, present strategies from pure language processing have been adopted, similar to using sequence-based fashions (e.g. recurrent neural networks [18] and Transformers [19]) to deal with 1D and 2D constructions, or graph neural networks (GNNs) for dealing with 3D representations. There might be many simplified molecular-input line-entry specification (SMILES) strings that each one signify the identical graph, whereas a recurrent mannequin must be taught to map all legitimate permutations of the SMILES string to the identical output. A GNN, which inherently respects permutation symmetries, handles this extra elegantly.
The properties and interactions of a molecule usually are not solely based mostly on the interatomic bonds and the data supplied in a molecular graph, however are additionally decided by the underlying bodily interactions between the atoms, which depend upon their 3D geometry [20]. Due to this, 3D molecular representations encode the total geometry of the molecule and thus convey in additional info. That is particularly helpful when figuring out whether or not a molecule is the suitable match for a binding receptor on a cell membrane.
Together with 3D details about molecules as further enter to a machine studying mannequin is anticipated to end in a extra expressive and lifelike technique of defining their dynamics. The sector of geometric deep studying (GDL) permits us to leverage the intrinsic geometrical options of a molecule and improve the standard of our fashions. Lately, GDL has repeatedly confirmed to be helpful in producing insights on related molecular properties, on account of its pure characteristic extraction capabilities, which will likely be mentioned beneath [6].
Symmetries, Chiralities, and Geometric Deep Studying
Bodily techniques and their interactions are inherently equivariant [21]: orientations inside the system shouldn’t change the bodily legal guidelines that govern the behaviour and properties of interacting components. Within the subject of GDL, symmetry is taken into account to be an indispensable matter, because it encompasses the properties of an arbitrary coordinate system with respect to transformations similar to translations, reflections, rotations, scalings, or permutations [17].
Conventional machine studying fashions make no assumption of symmetry (and therefore, usually are not symmetry-aware), and are delicate to the selection of coordinate system. To have the ability to acknowledge an n-dimensional sample in any orientation, such a mannequin must explicitly be handed each doable configuration and permutation of an enter (similar to a picture) to grasp all enter instances. To keep away from having to enhance the info a mess of occasions, one might leverage the geometry of the picture. Leveraging the geometry of the duty at hand just isn’t a novel idea — within the subject of picture evaluation, when contemplating a 2-D picture, convolutional neural networks (CNNs) are in a position to detect an object regardless of its place inside the picture. By respecting the geometry of the duty at hand, the mannequin learns extra effectively, as one can considerably cut back the variety of trainable parameters [22].
In machine studying, the notion of symmetry is commonly recast by way of invariance and equivariance. These specific the behaviour of any mathematical perform with respect to a change $T$ of an appearing symmetry group [23]. Primarily based on the definitions supplied in [17], we illustrate these ideas beneath. We take a mathematical perform, $F$, which we apply to a given enter, $X$. $F(X)$ can therein remodel equivariantly or invariantly:
- Equivariance: A perform $F$ utilized to an enter $X$ is equivariant to a change $T$ if the transformation of $X$ commutes with the transformation of $F(X)$, through a change $T$ of the identical symmetry group, such that: $F(T(X)) = T(F(X))$.
- Invariance: Invariance is a particular case of equivariance, the place $F(X)$ is invariant to $T$ if $T$ is the trivial group motion (i.e., identification): $F(T(X)) = T(F(X)) = F(X)$.
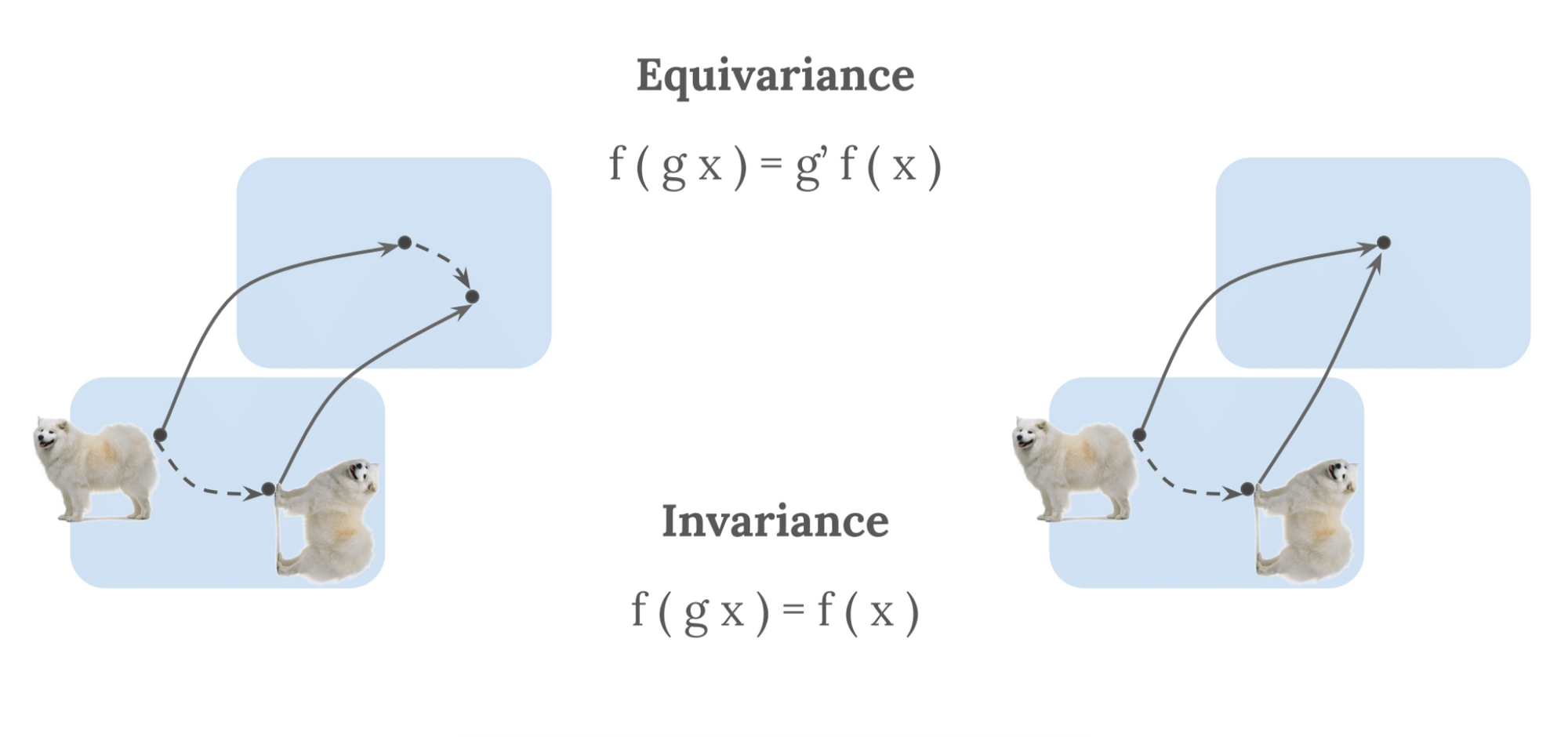
Or, in different phrases, equivariance offers with the notion that one transforms the output in the identical means one transforms the enter, whereas invariance offers with the notion that transformations of the enter don’t have an effect on the output, as seen in Determine 3.
For molecules, related transformations inside the 3D house contain rotations, and translations are a part of a symmetry group underneath which a system is invariant. This symmetry group is denoted because the particular Euclidean group in 3 dimensions, SE(3). The distinction between the Euclidean group in 3 dimensions, E(3), is the omission of the reflection as a legitimate transformation. This precept introduces us to the notion of translation invariance: for a molecule of atoms, translating our enter coordinates $r_1, dots, r_n$ by $g in R_3$ to change into $r_{1} +g, dots, r_{n} +g$, which shouldn’t change the output of the mannequin [20].
Nonetheless, this isn’t solely true for chiral molecules, as remodeling the molecular illustration does change the molecule. Their organic and pharmacological properties can considerably change relying on the handedness of the molecule. That is exemplified by enantiomers, that are pairs of molecules which might be mirror photographs of one another, however have totally different pharmacological properties [25].
Enantiomers can have very totally different results — mirroring an enantiomer might both yield a lead candidate or a lethal toxin [26]. With this in thoughts, it will be fascinating to contemplate machine studying techniques that keep in mind the symmetry of the duty at hand. Thus, integrating symmetry into these modelling duties has just lately been thought of a useful addition to the mannequin to signify these techniques and interactions extra faithfully. Some latest papers have leveraged this method inside the subject of molecular design, of which two will likely be briefly defined beneath.
E(n) Equivariant Normalizing Flows
To know the functioning of the normalizing stream, we first begin by contemplating the category of generative fashions. Underneath the umbrella time period of generative fashions, we take into account (1) energy-based fashions, (2) variational autoencoders, (3) generative adversarial networks, (4) normalizing flows, and (5) diffusion fashions.
Mathematically outlined, generative fashions usually encompass two components, that are a distribution $p_z$ and a learnable perform $g: z rightarrow x$, the place $z$ denotes the latent house and $x$ denotes an arbitrary level within the pattern house. Discriminative fashions, alternatively, have a learnable perform of the shape $g: x rightarrow y$, the place $x$ denotes an arbitrary level within the pattern house, and $y$ denotes the labels of the discovered options within the characteristic house. The distinction lies in how $g$ is discovered, i.e. what the target perform is. Generative fashions intention to mannequin the coaching distribution to be able to pattern new knowledge factors, while discriminative fashions intention to mannequin the choice boundary between courses.
Whereas the aforementioned generative fashions all share distinct aims, they might not essentially be taught the precise chance density perform $p(x)$ and chance, that are each tough to compute. That is the place the category of normalizing flows are available in. The chance of some extent $x$ might be discovered by inverting the learnable perform $g^{-1}$ and going again to some extent within the latent house $z$, which is finished via a slight change within the variables. Making use of normalizing flows to molecular situations, on this case, producing molecular options and 3-D positions, would require the positional info of nodes $x in R^{Mtimes n}$, the place $M$ denotes the variety of nodes current within the $n$-dimensional house. An atom sort is represented by $h$, the place $h$ is the illustration in $R^{M occasions nf}$. The discrete options $h$ are mapped right into a steady latent house, yielding the next for the change of variables system [27,28].
The learnable perform $f$ is constructed by the event of a Neural Odd Differential Equation (NODE) of which the dynamics are ruled by an E(n) Equivariant Graph Neural Community (E-GNN) [29]. In differential equations, we’d merely intention to seek out the by-product of a perform. Nonetheless, extraordinary differential equations use the idea of evolution of the by-product that slowly converges in direction of an analytical estimate of the answer. Integrating the NODE collects all of the phrases for the chance, amongst which lies the bottom distribution on the output $log P_z(z_x, z_h)$ [28]. Taken all collectively, this is able to yield the next process:
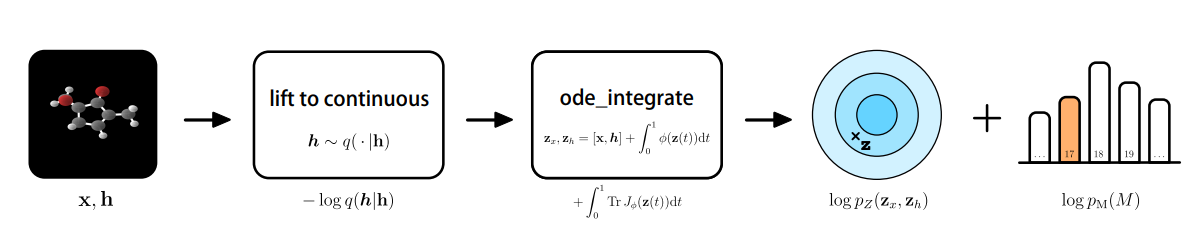
It was discovered that the E-NF considerably outperformed non-equivariant fashions by way of log-likelihood and that it produced 85% legitimate atom configurations based mostly on 10.000 generated examples. Utilizing the tactic, 4.91% had been steady, and of this subset, 99.80% had been distinctive and 93.28% had been novel.
E(3) Equivariant Diffusion Fashions
A newer technique is using E(3) Equivariant Diffusion Fashions for molecule technology [30,31]. Diffusion fashions fall underneath the identical class of generative fashions, which have been described above. Nonetheless, the sampling process differs. Allow us to recall that now we have the positional info $x$ of the nodes and $h$ denoting the totally different atom varieties. A ahead diffusion course of is outlined by including a small quantity of Gaussian noise to the pattern in $T$, producing a sequence of noisy samples $x_1, dots, x_t$, with a managed step dimension [32]. In different phrases, diffusion fashions are educated to reverse this noising course of. With a purpose to generate new samples at inference time, one begins with a Gaussian distribution and makes use of the educated diffusion mannequin to take away noise stepwise till one arrives at a “last” pattern. Evaluating this method to the earlier technique, it was discovered that it considerably outperforms state-of-the-art generative strategies by way of the scalability, high quality, and effectivity of the community. Bringing this collectively yields the next process:
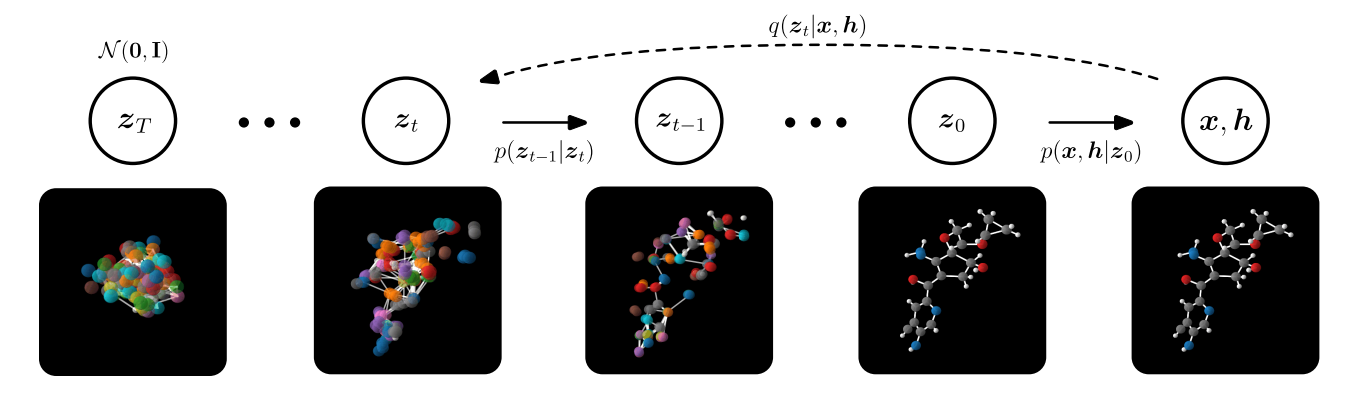
Compared to the E-NF, EDMs are in a position to generate 16x extra steady molecules than E-NF, whereas solely utilizing half of the coaching time, which considerably will increase the quantity of (novel) compounds accessible. The extra steady and testable compounds there are, the upper the chance of discovering a novel candidate within the high-throughput screening section which may work. Nonetheless, what’s necessary to notice is that stability is a trivial metric for in silico design as all drug molecules have to be steady. Nonetheless, a extra related metric could be synthesizability (which refers back to the steps wanted to synthesise the molecule) as a result of many steady, generated molecules are nowhere close to synthesizable [33].
Nonetheless, if we put the offered numbers from each research in perspective — typical success charges (so-called “hit-rates”, which showcase probably the most optimum molecule based mostly on their bioactivity) from human-based experimental strategies are within the vary of 0.01-0.14%, whereas digital display screen charges are between 1-40% [34], exhibiting a big enhance find one of the best compounds. It have to be famous that the experiments described in each of the research are carried out on datasets with a small variety of samples.
On the scale of real-world purposes, integrating the total geometric info would require an unimaginable quantity of computational assets. Options have been proposed, for instance in [20], which gives a novel method to implicitly integrating the 3D geometry of molecules into the neural community’s studying course of, whereas solely requiring 2D molecular graphs when utilizing the community to make predictions. Whereas utilizing extra detailed representations (i.e. 2D -> 3D -> past) is a step in the suitable route, the provision of information drastically drops. The true-world efficiency of those fashions is bottlenecked by the provision of high-quality geometric knowledge (not like the benchmark datasets in tutorial papers). To make additional progress, we have to begin serious about knowledge technology. Regardless that the latest strategies have demonstrated their success in small-scale case research, and there is likely to be extra on the horizon — this would possibly simply be a glimpse of the undiscovered iceberg of future drug candidates.
A New Period of Scientific Discovery?
We’ve got seen how machine studying can speed up the method of conventional drug discovery, and it’s simple that it’ll act as a robust catalyst for future analysis.
Utilizing computer-aided strategies for drug discovery considerably reduces the financial and human assets spent on the screening process, as they will discover appropriate lead candidates quicker, and commit much less time to synthesising non-promising compounds. This aids us in our understanding of how drug candidates work, as these fashions add an additional layer to the preclinical drug discovery funnel. This brings us to the prospect of reducing prices for drug improvement, with the eventual grand goal to develop medicine for uncommon illnesses. This paradigm shift can doubtlessly complement our present organic and chemical understanding of the pure and bodily sciences.
The latest advances in AI have nice potential to reinvent the established order and rethink and speed up how we deal with the various international, societal challenges we face. It’s simple that their affect will solely enhance within the coming years. With the appearance of Moore’s Legislation in 1965, which has been thought of a driving power of technological innovation and social change, this isn’t going to vary anytime quickly. What was unimaginable generations in the past turns into conceivable. Molecules are important to life as we all know it. They carry info to our cells from different cells, our meals, the air we breathe, and the medicine we take. Their complexity signifies that they’re each basic to biology while nonetheless being poorly understood. Nonetheless, advances in our understanding of the physicochemical properties of molecules and the way these properties work together with cells are anticipated to usher in a brand new period of drug discovery. It is extremely doable that the period has simply begun.
Creator Bio
Meilina Reksoprodjo is an MSc pupil in Information Science in Engineering on the Eindhoven College of Know-how (TU/e), specializing in explainable AI & geometric deep studying within the context of drug discovery. She holds a BSc diploma in Synthetic Intelligence from Radboud College. For extra updates, she is lively on Twitter or Linkedin.
Acknowledgements
The writer is grateful to Marco Cognetta, David D, Lin Min Htoo, Koen Minartz, Rıza Ozcelik, Tadija Radusinović, Maxim Snoep, Dr. Namid Stillman, Esther Shi, Koen Smeets, Sang Truong, Joost van der Haar, Stefan van der Sman, Derek van Tilborg and Dr. Yew Mun Yip for proofreading this text and offering useful enter for bettering the textual content.
Quotation
For attribution in tutorial contexts or books, please cite this work as
Meilina Reksoprodjo, “Symmetries, Scaffolds, and a New Period of Scientific Discovery”, The Gradient, 2022.
BibTeX quotation:
@article{reksoprodjooverview2022,
writer = {Meilina Reksoprodjo},
title = {Symmetries, Scaffolds, and a New Period of Scientific Discovery},
journal = {The Gradient},
12 months = {2022},
howpublished = {url{https://thegradient.pub/symmetries-scaffolds-and-a-new-era-of-scientific-discovery} },
}
Bibliography
[1] D. Solar, W. Gao, H. Hu and S. Zhou, “Why 90% of scientific drug improvement fails and how you can enhance it?”, Acta Pharmaceutica Sinica B, vol. 12, no. 7, pp. 3049-3062, 2022. Obtainable: 10.1016/j.apsb.2022.02.002.
[2] A. Mullard, “Parsing scientific success charges”, Nature Opinions Drug Discovery, vol. 15, no. 7, pp. 447-447, 2016. Obtainable: 10.1038/nrd.2016.136.
[3] R. Gómez-Bombarelli et al., “Automated Chemical Design Utilizing a Information-Pushed Steady Illustration of Molecules”, ACS Central Science, vol. 4, no. 2, pp. 268-276, 2018. Obtainable: 10.1021/acscentsci.7b00572.
[4] J. Kim and A. Scialli, “Thalidomide: The Tragedy of Beginning Defects and the Efficient Remedy of Illness”, Toxicological Sciences, vol. 122, no. 1, pp. 1-6, 2011. Obtainable: 10.1093/toxsci/kfr088.
[5] C. Yerkes, “Lecture 22”, Butane.chem.uiuc.edu, 2022. [Online]. Obtainable: http://butane.chem.uiuc.edu:80/cyerkes/Chem204sp06/Lecture_Notes/lect21c.html.
[6] M. M. Bronstein, J. Bruna, T. Cohen, and P. Velickovič, “Geometric deep studying: Grids, teams, graphs, geodesics, and gauges”, arXiv:2104.13478, 2021.
[7] J. Vamathevan et al., “Functions of machine studying in drug discovery and improvement”, Nature Opinions Drug Discovery, vol. 18, no. 6, pp. 463-477, 2019. Obtainable: 10.1038/s41573-019-0024-5.
[8] G. Sliwoski, S. Kothiwale, J. Meiler and E. Lowe, “Computational Strategies in Drug Discovery”, Pharmacological Opinions, vol. 66, no. 1, pp. 334-395, 2013. Obtainable: 10.1124/pr.112.007336.
[9] S. Kim et al., “PubChem Substance and Compound databases”, Nucleic Acids Analysis, vol. 44, no. 1, pp. D1202-D1213, 2015. Obtainable: 10.1093/nar/gkv951.
[10] G. Papadatos, A. Gaulton, A. Hersey and J. Overington, “Exercise, assay and goal knowledge curation and high quality within the ChEMBL database”, Journal of Laptop-Aided Molecular Design, vol. 29, no. 9, pp. 885-896, 2015. Obtainable: 10.1007/s10822-015-9860-5.
[11] J. Howard, “The enterprise influence of deep studying”, Proceedings of the nineteenth ACM SIGKDD worldwide convention on Information discovery and knowledge mining, 2013. Obtainable: 10.1145/2487575.2491127.
[12] B. McMahon, “AI is Ushering In a New Scientific Revolution”, The Gradient, 2022. Obtainable: https://thegradient.pub/ai-scientific-revolution/
[13] H. Chen, O. Engkvist, Y. Wang, M. Olivecrona and T. Blaschke, “The rise of deep studying in drug discovery”, Drug Discovery At present, vol. 23, no. 6, pp. 1241-1250, 2018. Obtainable: 10.1016/j.drudis.2018.01.039.
[14] Ok. Roy, S. Kar and R. Das, A primer on QSAR/QSPR modeling, 1st ed. Springer, 2015, pp. 1-36.
[15] Z. Wu et al., “MoleculeNet: a benchmark for molecular machine studying”, Chemical Science, vol. 9, no. 2, pp. 513-530, 2018. Obtainable: 10.1039/c7sc02664a.
[16] D. Weininger, “SMILES, a chemical language and knowledge system. 1. Introduction to methodology and encoding guidelines”, Journal of Chemical Data and Modeling, vol. 28, no. 1, pp. 31-36, 1988. Obtainable: 10.1021/ci00057a005.
[17] Ok. Atz, F. Grisoni and G. Schneider, “Geometric deep studying on molecular representations”, Nature Machine Intelligence, vol. 3, no. 12, pp. 1023-1032, 2021. Obtainable: 10.1038/s42256-021-00418-8.
[18] E. Jannik Bjerrum, R. Threlfall. “Molecular Era with Recurrent Neural Networks (RNNs)”. arXiv:1705.04612, 2017.
[19] Y. Rong et al., “Self-Supervised Graph Transformer on Massive-Scale Molecular Information”, Proceedings of the 33th Worldwide Convention on Neural Data Processing Programs (NeurIPS), 2020, pp. 12559–12571.
[20] H. Stark et al., “3-D Infomax improves GNNs for Molecular Property Prediction”, offered on the thirty ninth Worldwide Convention on Machine Studying (ICML), 2022.
[21] T. Smidt, “Euclidean Symmetry and Equivariance in Machine Studying”, Tendencies in Chemistry, vol. 3, no. 2, pp. 82-85, 2021. Obtainable: 10.1016/j.trechm.2020.10.006.
[22] A. White, “Deep studying for molecules and supplies”, Residing Journal of Computational Molecular Science, vol. 3, no. 1, 2022. Obtainable: 10.33011/livecoms.3.1.1499.
[23] J. Marsden and A. Weinstein, “Discount of symplectic manifolds with symmetry”, Studies on Mathematical Physics, vol. 5, no. 1, pp. 121-130, 1974. Obtainable: 10.1016/0034-4877(74)90021-4.
[24] E. van der Pol, “AMMI Seminar 1 – Geometric Deep Studying and Reinforcement Studying”, African Masters of Machine Intelligence [Online], 2021.
[25] L. Nguyen, H. He and C. Pham-Huy, “Chiral Medication: An Overview”, Worldwide Journal of Biomedical Science, vol. 2, no. 2, pp. 85-100, 2006.
[26] W. H. Brooks, W. C. Guida and Ok. G. Daniel, “The Significance of Chirality in Drug Design and Growth”, Present Subjects in Medicinal Chemistry, vol. 11, no. 7, pp. 760-770, 2011. Obtainable: 10.2174/156802611795165098.
[27] V. G. Satorras, E. Hoogeboom, F. B. Fuchs, I. Posner, and M. Welling. “E(n) equivariant Normalizing Flows”, within the thirty fourth Advances in Neural Data Processing Programs (NeurIPS) [Online], 2021. Obtainable: https://proceedings.neurips.cc/paper/2021/hash/21b5680d80f75a616096f2e791affac6-Abstract.html
[28] E. Hoogeboom, “Find out how to construct E(n) Equivariant Normalizing Flows, for factors with options?”, ehoogeboom.github.io/, 2022. [Online]. Obtainable: https://ehoogeboom.github.io/put up/en_flows/.
[29] V. G. Satorras, E. Hoogeboom, and M. Welling. “E(n) equivariant graph neural networks”, in Proceedings of the thirty eighth Worldwide Convention on Machine Studying (ICML) [Online], 2021. Obtainable: http://proceedings.mlr.press/v139/satorras21a/satorras21a.pdf.
[30] E. Hoogeboom et al., “Equivariant Diffusion for Molecule Era in 3-D”, in Proceedings of the thirty ninth Worldwide Convention on Machine Studying,(ICML), 2022, pp. 8867–8887.
[31] M. Xu, L. Yu, Y. Track, C. Shi, S. Ermon and J. Tang, “GeoDiff: A Geometric Diffusion Mannequin for Molecular Conformation Era”, offered on the eleventh Worldwide Convention on Studying Representations (ICLR 2022) [Online]. Obtainable: arXiv:2203.02923.
[32] L. Weng, “What are Diffusion Fashions?”, lilianweng.github.io, 2022. [Online]. Obtainable: https://lilianweng.github.io/posts/2021-07-11-diffusion-models/.
[33] W. Gao and C. Coley, “The Synthesizability of Molecules Proposed by Generative Fashions”, Journal of Chemical Data and Modeling, vol. 60, no. 12, pp. 5714-5723, 2020. Obtainable: 10.1021/acs.jcim.0c00174.
[34] T. Zhu et al., “Hit Identification and Optimization in Digital Screening: Sensible Suggestions Primarily based on a Important Literature Evaluation”, Journal of Medicinal Chemistry, vol. 56, no. 17, pp. 6560-6572, 2013. Obtainable: 10.1021/jm301916b.
[ad_2]
Source link
